共识基序搜索#
![]()
本教程利用了Matrix Profile XV 论文的主要要点。
矩阵剖面 可以用于 在单个时间序列中找到保留的模式 (自连接) 和 在两个时间序列之间 (AB连接)。在这两种情况下,这些保留的模式通常称为“主题”。而且,当考虑三条或更多时间序列时,识别整个集合中保留主题的一个常见技巧是:
在每个时间序列的末尾附加一个
np.nan。这用于识别相邻时间序列之间的边界,并确保任何识别出的模式不会跨越多个时间序列。将所有时间序列连接成一个单一的长时间序列
计算上述连接时间序列的矩阵轮廓(自连接)
然而,这并不能保证找到在集合中所有时间序列中都保留的模式。这个在一组时间序列中找到一个共同保留的动机的想法被称为“共识动机”。在本教程中,我们将介绍“Ostinato”算法,这是一种在一组时间序列中高效找到共识动机的方法。
开始使用#
让我们导入需要加载、分析和绘制数据的包。
%matplotlib inline
import stumpy
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
from matplotlib.patches import Rectangle
from scipy.cluster.hierarchy import linkage, dendrogram
plt.style.use('https://raw.githubusercontent.com/TDAmeritrade/stumpy/main/docs/stumpy.mplstyle')
加载眼动追踪 (EOG) 数据集#
在以下数据集中,一名志愿者被要求通过执行代表个别日文字符书写笔画的眼动来“拼写”不同的日文句子。他们的眼动通过电眼动仪(EOG)记录,并被给予一秒钟的时间来“视觉描摹”每个日文字符。对于我们的目的,我们仅使用垂直眼位,并且从概念上讲,这个基本示例再现了Matrix Profile XV论文的图1和图2。
Ts = []
for i in [6, 7, 9, 10, 16, 24]:
Ts.append(pd.read_csv(f'https://zenodo.org/record/4288978/files/EOG_001_01_{i:03d}.csv?download=1').iloc[:, 0].values)
可视化EOG数据集#
下面,我们绘制了六个时间序列,每个序列代表一个人在用眼睛“书写”日语句子时的垂直眼位。正如您所看到的,有些日语句子较长,包含更多的单词,而另一些则较短。然而,有一个共同的日语单词(即,“共同主题”)包含在所有六个示例中。您能找出在这六个时间序列中共有的、持续一秒钟的模式吗?
def plot_vertical_eog():
fig, ax = plt.subplots(len(Ts), sharex=True, sharey=True)
colors = plt.rcParams["axes.prop_cycle"]()
for i, T in enumerate(Ts):
ax[i].plot(T, color=next(colors)["color"])
ax[i].set_ylim((-330, 1900))
plt.subplots_adjust(hspace=0)
plt.xlabel('Time')
return ax
plot_vertical_eog()
plt.suptitle('Vertical Eye Position While Writing Different Japanese Sentences', fontsize=14)
plt.show()

共识基序搜索#
要找到答案,我们可以使用 stumpy.ostinato 函数,通过传入时间序列列表 Ts 以及子序列窗口大小 m 来帮助我们发现“共识特征”。
m = 50 # Chosen since the eog signal was downsampled to 50 Hz
radius, Ts_idx, subseq_idx = stumpy.ostinato(Ts, m)
print(f'Found Best Radius {np.round(radius, 2)} in time series {Ts_idx} starting at subsequence index location {subseq_idx}.')
Found Best Radius 0.87 in time series 4 starting at subsequence index location 1271.
现在,让我们绘制与匹配的共识基序相对应的每个时间序列的单独子序列:
consensus_motif = Ts[Ts_idx][subseq_idx : subseq_idx + m]
nn_idx = []
for i, T in enumerate(Ts):
nn_idx.append(np.argmin(stumpy.core.mass(consensus_motif, T)))
lw = 1
label = None
if i == Ts_idx:
lw = 4
label = 'Consensus Motif'
plt.plot(stumpy.core.z_norm(T[nn_idx[i] : nn_idx[i]+m]), lw=lw, label=label)
plt.title('The Consensus Motif (Z-normalized)')
plt.xlabel('Time (s)')
plt.legend()
plt.show()
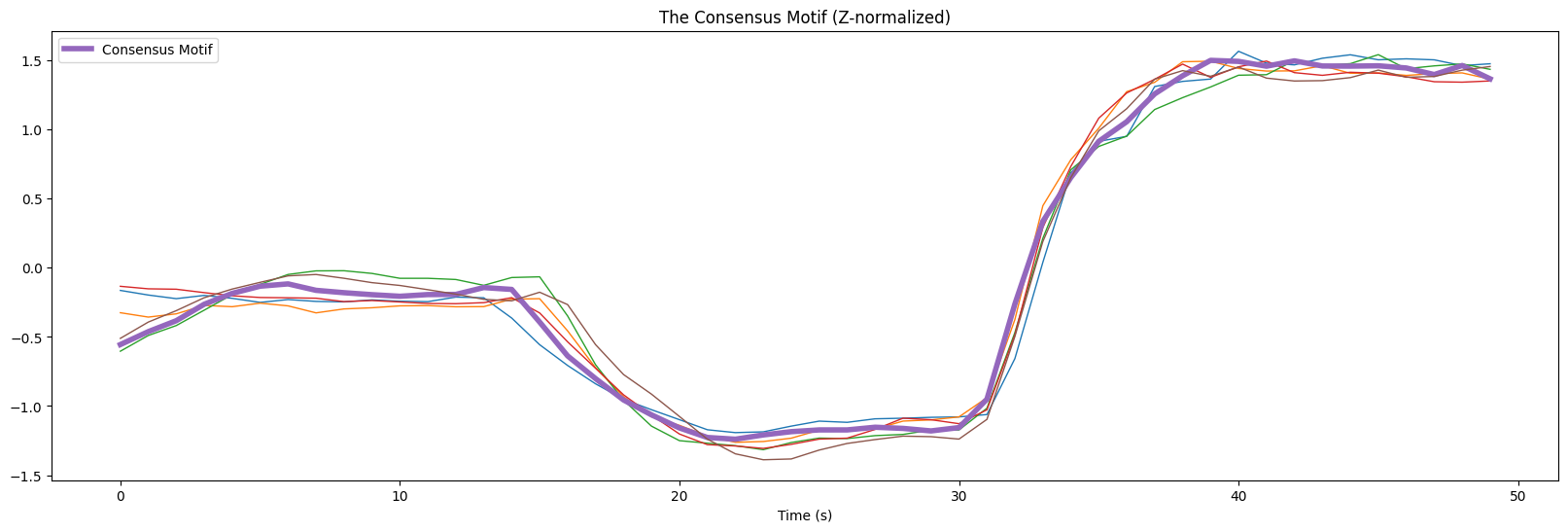
子序列之间存在显著的相似性。最中心的“共识特征”用更粗的紫色线条绘制。
当我们在其原始上下文中突出显示上述子序列(下方浅蓝色框)时,我们可以看到它们发生在不同的时间:
ax = plot_vertical_eog()
ymin, ymax = ax[i].get_ylim()
for i in range(len(Ts)):
r = Rectangle((nn_idx[i], ymin), m, ymax-ymin, alpha=0.3)
ax[i].add_patch(r)
plt.suptitle('Vertical Eye Position While Writing Different Japanese Sentences', fontsize=14)
plt.show()
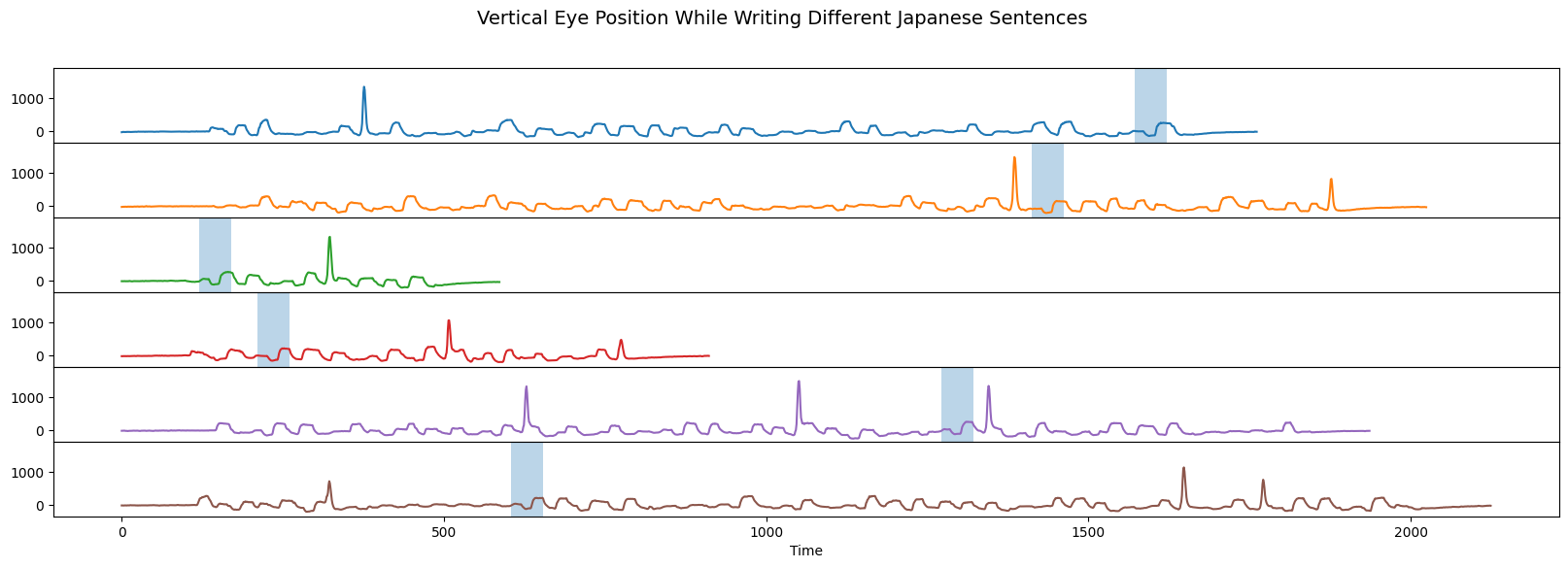
发现的保守基序(浅蓝色框)对应于书写日本字符 ア,它在不同的例子句中出现的时间不同。
使用线粒体DNA (mtDNA) 的系统发育#
在下一个例子中,我们将重现Matrix Profile XV论文中的图9。
线粒体DNA (mtDNA) 已成功用于确定生物之间的进化关系(系统发育)。由于DNA本质上是字母的有序序列,我们可以将它们松散地视为时间序列,并使用所有可用的时间序列工具。
加载mtDNA数据集#
animals = ['python', 'hippo', 'red_flying_fox', 'alpaca']
dna_seqs = {}
truncate = 15000
for animal in animals:
dna_seqs[animal] = pd.read_csv(f"https://zenodo.org/record/4289120/files/{animal}.csv?download=1").iloc[:truncate, 0].values
colors = {'python': 'tab:blue', 'hippo': 'tab:green', 'red_flying_fox': 'tab:purple', 'alpaca': 'tab:red'}
使用大型mtDNA序列进行聚类#
天真地使用 scipy.cluster.hierarchy 我们可以根据大多数序列对mtDNA进行聚类。一个正确的聚类会将两个“偶蹄目”——河马和美洲驼——放在最接近的位置,并且,连同红飞狐,我们预期它们会形成一个“哺乳动物”的聚类。最后,python作为一种“爬行动物”,应该距离所有的“哺乳动物”最远。
fig, ax = plt.subplots(ncols=2)
# Left
for animal, dna_seq in dna_seqs.items():
ax[0].plot(dna_seq, label=animal, color=colors[animal])
ax[0].legend()
ax[0].set_xlabel('Number of mtDNA Base Pairs')
ax[0].set_title('mtDNA Sequences')
# Right
pairwise_dists = []
for i, animal_1 in enumerate(animals):
for animal_2 in animals[i+1:]:
pairwise_dists.append(stumpy.core.mass(dna_seqs[animal_1], dna_seqs[animal_2]).item())
Z = linkage(pairwise_dists, optimal_ordering=True)
dendrogram(Z, labels=animals, ax=ax[1])
ax[1].set_ylabel('Z-normalized Euclidean Distance')
ax[1].set_title('Clustering')
plt.show()
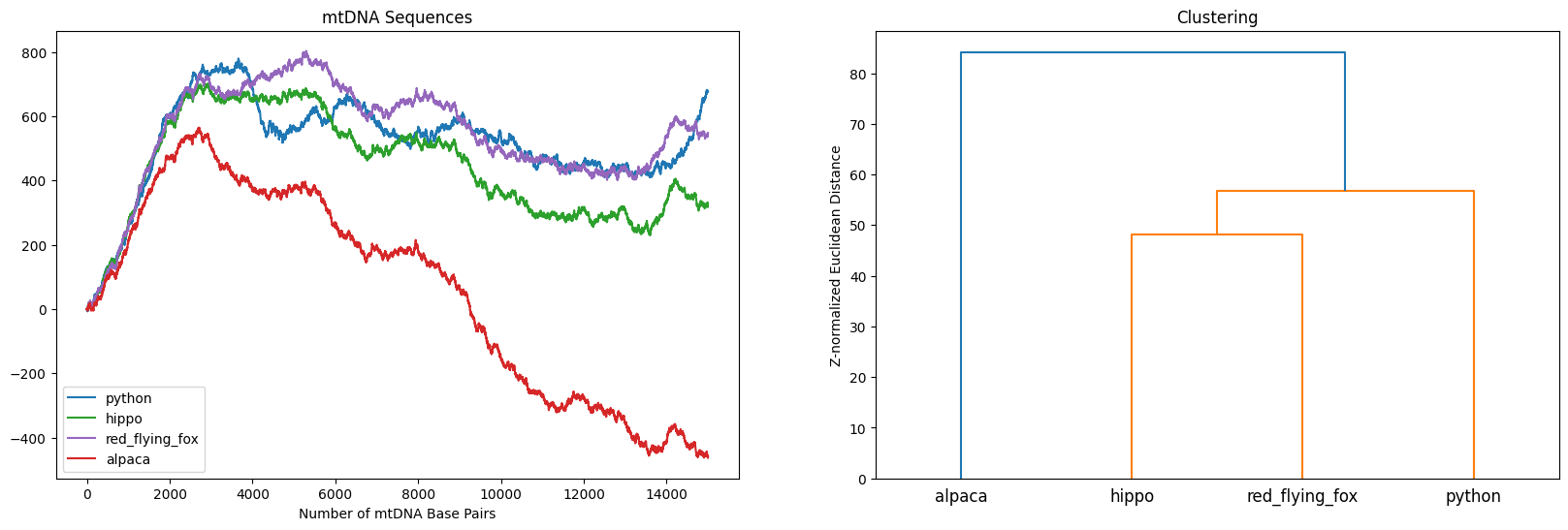
哎呀,聚类显然是错误的!除了其他问题外,羊驼(一种哺乳动物)不应该与蟒蛇(一种爬行动物)最为相关。
共识基序搜索#
为了获得正确的关系,我们需要识别并比较mtDNA序列中保存最好的部分。换句话说,我们需要基于它们的共识基序进行聚类。让我们将子序列窗口大小限制为1,000个碱基对,并再次使用stumpy.ostinato函数识别共识基序:
m = 1000
radius, Ts_idx, subseq_idx = stumpy.ostinato(list(dna_seqs.values()), m)
print(f'Found best radius {np.round(radius, 2)} in time series {Ts_idx} starting at subsequence index location {subseq_idx}.')
Found best radius 2.73 in time series 1 starting at subsequence index location 602.
使用共识mtDNA基序进行聚类#
现在,让我们再次进行聚类,但这次使用共识特征:
consensus_motif = list(dna_seqs.values())[Ts_idx][subseq_idx : subseq_idx + m]
# Extract Animal DNA Subsequence Closest to the Consensus Motif
dna_subseqs = {}
for animal in animals:
idx = np.argmin(stumpy.core.mass(consensus_motif, dna_seqs[animal]))
dna_subseqs[animal] = stumpy.core.z_norm(dna_seqs[animal][idx : idx + m])
fig, ax = plt.subplots(ncols=2)
# Left
for animal, dna_subseq in dna_subseqs.items():
ax[0].plot(dna_subseq, label=animal, color=colors[animal])
ax[0].legend()
ax[0].set_title('Consensus mtDNA Subsequences (Z-normalized)')
ax[0].set_xlabel('Number of mtDNA Base Pairs')
# Right
pairwise_dists = []
for i, animal_1 in enumerate(animals):
for animal_2 in animals[i+1:]:
pairwise_dists.append(stumpy.core.mass(dna_subseqs[animal_1], dna_subseqs[animal_2]).item())
Z = linkage(pairwise_dists, optimal_ordering=True)
dendrogram(Z, labels=animals, ax=ax[1])
ax[1].set_title('Clustering Using the Consensus mtDNA Subsequences')
ax[1].set_ylabel('Z-normalized Euclidean Distance')
plt.show()
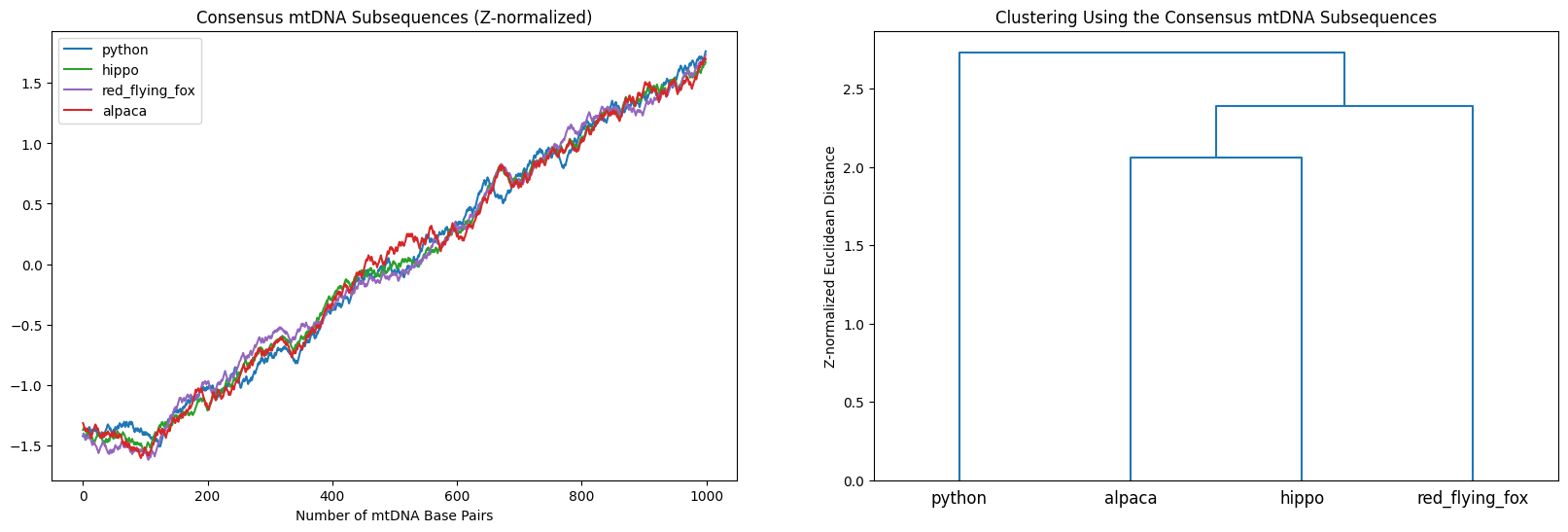
现在这看起来好多了!从层次上看,蟒蛇与其他哺乳动物“相距甚远”,在哺乳动物中,红飞狐(一种蝙蝠)与美洲驼和河马的关系较远,而美洲驼和河马是这组动物中最近的进化亲属。
总结#
就这样!您现在已经学习了如何在一组时间序列中搜索共识图案,使用了优秀的 stumpy.ostinato 函数。您现在可以导入这个包并在自己的项目中使用。祝您编码愉快!